Spatially-informed differential gene expression
2025-04-07
spatial_differential_expression.RmdDifferential gene expression testing is a fundamental part in studies
using “bulk” RNAseq data, and continues to be critical in the analysis
of single-cell and spatial transcriptomics (ST). The STdiff
function from the spatialGE
package provides methods for traditional, non-spatial differential gene
expression analysis such as Wilcoxon’s rank test, T-test, and mixed
models.
Spatial transcriptomics presents a new challenge for differential gene expression analysis. In ST, gene expression is measured at locations (spots or cells) that are separated by fixed or variable distances. Within each assayed tissue, cells or spots that are close are more likely to be transcriptomically similar compared to distant cells or spots. This correlation between transcriptomic similarity and spatial distance is known as spatial dependency. Neglecting the spatial dependency in ST may result in inflation of type I error (Ospina et al. (2024)) and an excess of differentially expressed genes (false positives).
To visually illustrate the existence of spatial dependency in ST data, a variogram can be used. A variogram is a plot that shows how a variable (gene expression in this case), varies with respect to distance among the locations (spots or cells).
Spatial dependency in ST data (melanoma stage III tumor biopsies)
The spatial dependency is described quantitatively by means of the spatial covariance among spots/cells. The estimation of spatial covariance is computationally expensive, especially if the ST data that contains thousands of spots/cells. For convenience, in this tutorial the smaller ST data set generated by Thrane et al. (2018) will be used. This data set includes lymph node biopsies from four patients with stage III melanoma, with two tissue slices per biopsy. This technology is a predecessor of the Visium assay, and allowed RNA capture in up to 1,007 spots separated each other by 200μM. The spots are 100μM in diameter, which corresponds to 5-40 cells according to the authors.
For this tutorial, we will use two of the eight samples in Thrane et al. (2018). The data set is included in the spatialGE_Data GitHub repository repository, but users can also find the data in the authors’ website.
Creating an STlist (Spatial Transcriptomics List)
The spatialGE package does not have built-in methods to
generate variogram plots, however an STlist object will be created to
normalize the ST data, which is necessary for variogram analysis. The
same STlist will be used later to conduct differential expression
analysis with STdiff.
Load the spatialGE package:
library('spatialGE')The STlist function takes data in several formats. The
reader is encouraged to see the STlist reference (see here
to learn about the different options to read ST data in
spatialGE. In this guide, we provide the
STlist function with comma-delimited files containing gene
expression counts per spot and comma-delimited containing the spatial
coordinates per spot. The example files in this tutorial can be
downloaded from the spatialGE_Data
GitHub repository. Users are encouraged to download these files to
familirize themselves with the data format.
In this tutorial, data will be downloaded and deposited in a temporary directory. However, the download path can be changed within the following code block:
thrane_tmp = tempdir()
unlink(thrane_tmp, recursive=TRUE)
dir.create(thrane_tmp)
download.file('https://github.com/FridleyLab/spatialGE_Data/raw/refs/heads/main/melanoma_thrane.zip?download=',
destfile=paste0(thrane_tmp, '/', 'melanoma_thrane.zip'), mode='wb')
zip_tmp = list.files(thrane_tmp, pattern='melanoma_thrane.zip$', full.names=TRUE)
unzip(zipfile=zip_tmp, exdir=thrane_tmp)From the temporary directory, we can use R to generate the file paths
to be passed to the STlist function:
# Find expression and coordinates files within the temporary folder
count_files <- list.files(paste0(thrane_tmp, '/melanoma_thrane'), full.names=TRUE, pattern='counts')
coord_files <- list.files(paste0(thrane_tmp, '/melanoma_thrane'), full.names=TRUE, pattern='mapping')This melanoma ST data set also contains sample-level meta data. We will also provide this file to specify sample names:
# Find clinical data within folder
clin_file <- list.files(paste0(thrane_tmp, '/melanoma_thrane'), full.names=TRUE, pattern='clinical')We can load the data into an STlist object with:
melanoma <- STlist(rnacounts=count_files, spotcoords=coord_files, samples=clin_file, cores=2)
#> Found matrix data
#> Matching gene expression and coordinate data...
#> Converting counts to sparse matrices
#> Completed STlist!Note: If you receive a warning indicating that an input file has a incomplete final line, you can ignore it. Alternatively, modify the input files with a text editor by adding an empty line at the end of the files.
The melanoma object is an STlist that contains the count
data, spot coordinates, and clinical meta data.
melanoma
#> Spatial Transcriptomics List (STlist).
#> 4 spatial array(s):
#> ST_mel1_rep2 (293 ROIs|spots|cells x 16148 genes)
#> ST_mel2_rep1 (383 ROIs|spots|cells x 16831 genes)
#> ST_mel3_rep1 (256 ROIs|spots|cells x 15653 genes)
#> ST_mel4_rep2 (248 ROIs|spots|cells x 15991 genes)
#>
#> 9 variables in sample-level data:
#> patient, slice, gender, BRAF_status, NRAS_status, CDKN2A_status, survival, survival_months, RINTo normalize the ST data, the transform_data is
used:
melanoma <- transform_data(melanoma, cores=2)A variogram of gene expression in ST data
The expression of the gene COL1A1 will be used to illustrate spatial dependency in ST. The gene COL1A1 is one of the collagen genes, a key component of the extracellular matrix and the stroma compartment. The stroma is the region in a cancer tissue that surrounds and often supports the tumor cells. The variogram can show how its expression varies with respect to the distance between spots within the sample “ST_mel1_rep2”.
Load tidtverse for data manipulation:
Then, data frame is created with both gene COL1A1 gene expression and spatial coordinates from sample “ST_mel1_rep2”.
# Sample and gene to be used in example
samplename <- 'ST_mel1_rep2'
gene <- 'COL1A1'
# Prepare data with coordinate and expression data
df_vgm <- melanoma@spatial_meta[[samplename]] %>%
select(libname, xpos, ypos) %>%
left_join(., tibble(libname=colnames(melanoma@tr_counts[[samplename]]),
geneexpr=as.vector(melanoma@tr_counts[[samplename]][gene, ])), by='libname') %>%
column_to_rownames(var='libname')The data frame contains the spatial coordinates of each spot
(xpos, ypos) and transformed expression of
MLANA (geneexpr).
head(df_vgm)
#> xpos ypos geneexpr
#> x7x15 7 15 0.000000
#> x7x16 7 16 1.852142
#> x7x17 7 17 1.789264
#> x7x18 7 18 0.000000
#> x8x13 8 13 0.000000
#> x8x14 8 14 0.000000In a variogram, each point describes the variance in gene expression
(semiovariance) with respect to the distance between spots. If spatial
dependency is present trend should be observed wherein semiovariance
increases as the distance between spots increases. In other words,
variance between close spots is lower than variance between distant
spots. To calculate the variogram, the geoR package is
used:
#> variog: computing omnidirectional variogramNow, the variogram can be plotted:
plot(vgm, pch=16, cex=0.5, ylab='Semiovariance', xlab='Distance',
main=paste0('Variogram of ', gene, ' expression'))
text(c(2, 5, 14), c(0.65, 2.1, 0.65),
labels=c('Nugget','Sill', 'Range'),
col=c('purple', 'green', 'goldenrod1'))
clip(x1=-1, x2=12, y1=-1, y2=2.1)
abline(h=2, col='green', lwd=3, lty=2)
clip(x1=-1, x2=1, y1=-1, y2=2)
abline(h=0.75, col='purple', lwd=3, lty=2)
clip(x1=-1, x2=12.1, y1=-1, y2=2)
abline(v=12, col='goldenrod1', lwd=3, lty=2)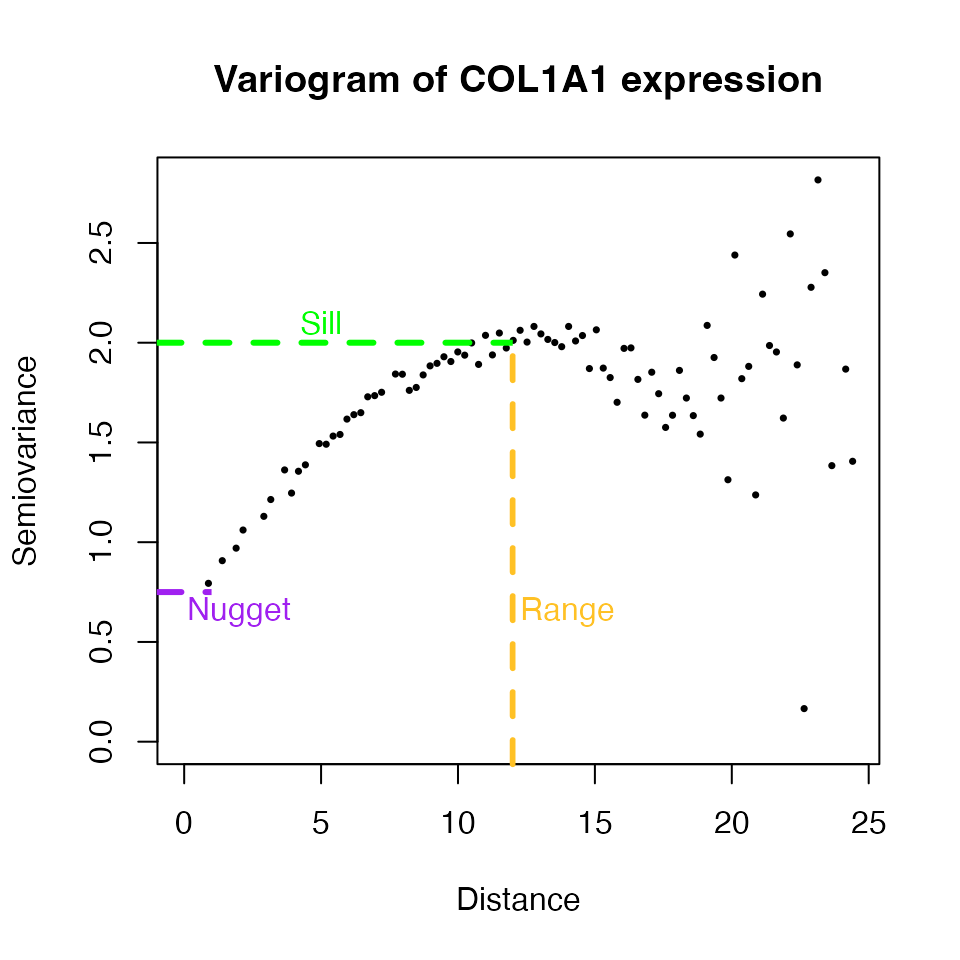
We can see that variance is higher at longer distances. Note, that this relationship is disrupted at longer spot-to-spot distances (> 15 in this case). This disruption occurs because as spot-to-spot distances increase, some spots may belong to distant tissue niches that hold similarity. Regardless, the positive relationship between COL1A1 expression variance and distance is clear a short to medium distances, first increasing and then forming a plateau. Since a relationship exists between COL1A1 expression variance and distance, spatial covariance models defined by the nugget, sill, and range parameters can be fitted to account for spatial dependency.
How is the information in variogram used by STdiff
?
To account for spatial dependency, the STdiff calculates
three parameters from the variogram that are necessary to describe the
relationship between gene expression variance and distance between spots
or cells. The three parameters are known as nugget,
sill, and range).
The spatial covariance can be modeled using different covariance structures. The covariance structure that best describes the relationship varies from gene to gene, and from sample to sample. Three popular spatial covariance structures are the exponential, spherical, and gaussian covariance structures, all of them defined in terms of the nugget, sill, and range parameters.
Using the variogram calculated previously, the different covariance structures can be fitted and visualized with geoR like so:
# Exponential covariance structure:
exp_model <- geoR::variofit(vgm, fix.nugget=FALSE, cov.model='exponential',
ini.cov.pars=c(0.2, 11), nugget=0.7, # Sill, range, and nugget specified here
max.dist=15) # Maximum distance to which model will be fitted
#> variofit: covariance model used is exponential
#> variofit: weights used: npairs
#> variofit: minimisation function used: optim
sph_model <- geoR::variofit(vgm, fix.nugget=FALSE, cov.model='spherical',
ini.cov.pars=c(0.2, 11), nugget=0.7,
max.dist=15)
#> variofit: covariance model used is spherical
#> variofit: weights used: npairs
#> variofit: minimisation function used: optim
gau_model <- geoR::variofit(vgm, fix.nugget=FALSE, cov.model='gaussian',
ini.cov.pars=c(0.2, 11), nugget=0.7,
max.dist=15)
#> variofit: covariance model used is gaussian
#> variofit: weights used: npairs
#> variofit: minimisation function used: optim
plot(vgm, pch=16, cex=0.5, ylab='Semiovariance', xlab='Distance',
main=paste0('Variogram of ', gene, ' expression with\nfitted covariance structures'))
text(c(20.5, 20.5, 20.5), c(0.8, 0.6, 0.4), labels=c('Exponential','Spherical', 'Gaussian'))
clip(x1=15, x2=17, y1=-1, y2=2.1)
abline(h=0.8, col=alpha('red', 0.7), lwd=3)
abline(h=0.6, col=alpha('blue', 0.7), lwd=3)
abline(h=0.4, col=alpha('orange', 0.7), lwd=3)
clip(x1=-1, x2=12, y1=-1, y2=2.1)
abline(h=2, col='green', lwd=3, lty=2)
clip(x1=-1, x2=1, y1=-1, y2=2)
abline(h=0.75, col='purple', lwd=3, lty=2)
clip(x1=-1, x2=12.1, y1=-1, y2=2)
abline(v=12, col='goldenrod1', lwd=3, lty=2)
lines(exp_model, col=alpha('red', 0.7), lwd=3)
lines(sph_model, col=alpha('blue', 0.7), lwd=3)
lines(gau_model, col=alpha('orange', 0.7), lwd=3)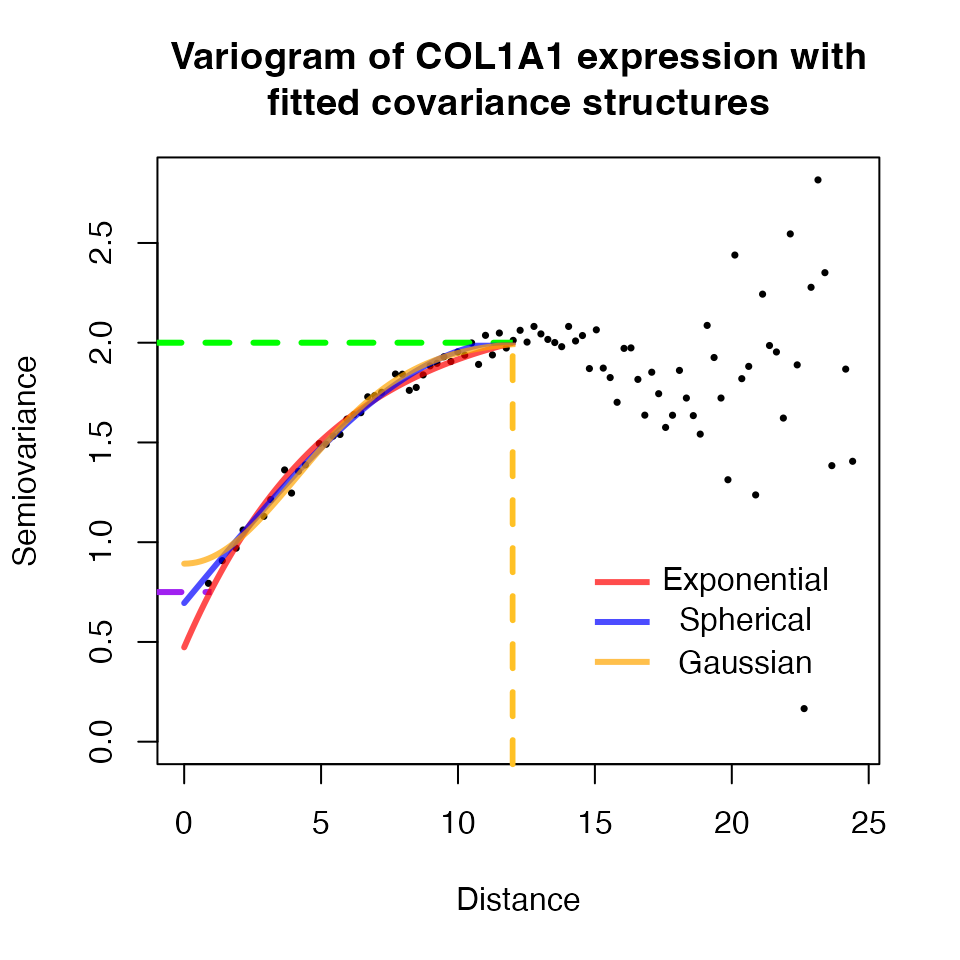
In the case of COL1A1 for sample “ST_mel1_rep2”, all models
(exponential, spherical, and gaussian) fit well the variogram. Due to
computational constraints, the geoR package is not used in
spatialGE to estimate the covariance structures. Rather,
the spaMM package has been used and only the exponential
covariance structure has been implemented. While this choice is not
optimal for all genes, accounting for the spatial dependency even if the
model fit is not optimal, is likely better than not accounting for the
spatial dependency at all.
Differential gene expression analysis with STdiff
In addition to Wilcoxon’s rank test and T-test, the
STdiff function can also use non-spatial mixed
models to test for differentially expressed genes. In the
non-spatial mixed models the the expression
of a gene in spot/cell
is described as:
where is the average expression of the gene in the sample, and is the random error at spot/cell .
To reduce the number of false positives in differential expression
analysis of ST data, the STdiff function features a novel
implementation of spatial mixed models with that expands the
non-spatial case to include spatial covariance structures. In the
spatial mixed models, the expression
of a gene in spot/cell
is described as:
where denotes the exponential spatial covariance of spot/cell .
In order to detect differentially expressed genes, groups of
spots/cells should be defined first. In the context of ST data, these
groups of spots/cells often represent tissue domains. The
spatialGE package includes a spatially-informed tissue
domain detection method called STclust.
Detection of tissue domains with STclust
The STclust function groups spots/cells into tissue
domains using hierarchical clustering on a wighted similarity matrix.
The similarity maxtrox is derived from Euclidean transcriptomics
distances “shrunk” by the spatial Euclidean distances. Please, refer to
the journal article describing the method for more details (Ospina et al. 2022). To detect tissue domains,
a spatial weight of 0.025 (w=0.025) will be used. The
number of domains will be automatically defined by applying
dynamicTreeClusters (ks='dtc').
melanoma <- STclust(melanoma,
ks='dtc',
ws=0.025, cores=2)
#> STclust started...
#> Updating STlist with results...
#> STclust completed in 0.03 min.Results of STclust domain detection can be plotted using
the STplot function:
cluster_p <- STplot(melanoma,
samples='ST_mel1_rep2',
ws=0.025,
deepSplit=FALSE,
ptsize=2, color_pal=c('#00BA38', '#EEBA38', '#619CFF', '#F8766D'))
print(cluster_p[[1]])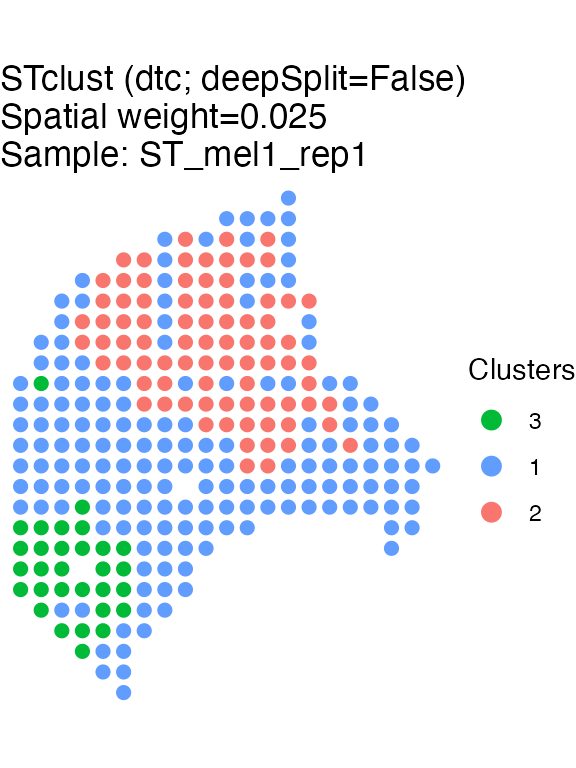
With the domains identified, the STdiff function can be
used. The following command tests for differentially expressed genes on
the 1000 genes with the highest variation (as defined by
Seurat’s function FindVariableFeatures).
deg <- STdiff(melanoma,
samples='ST_mel1_rep2',
w=0.025, k='dtc', deepSplit=FALSE,
topgenes=1000,
sp_topgenes=0.2, cores=2) # Test the 20% of the top differentially expressed genes (based on adjusted p-value)
#> Testing STclust assignment (w=0.025, dtc deepSplit=False)...
#> Running non-spatial mixed models...
#> Registered S3 methods overwritten by 'registry':
#> method from
#> print.registry_field proxy
#> print.registry_entry proxy
#> Completed non-spatial mixed models (3.22 min).
#> Running spatial tests...
#>
#> Completed spatial mixed models (1.39 min).
#> STdiff completed in 4.64 min.The output of STdiff is a list, where each element is a
data frame containing the test results for each sample:
print(deg[['ST_mel1_rep2']])
#> # A tibble: 3,000 × 9
#> sample gene avg_log2fc cluster_1 mm_p_val adj_p_val exp_p_val exp_adj_p_val
#> <chr> <chr> <dbl> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 ST_mel… GNAS -0.715 1 2.11e-15 4.36e-14 1.44e-9 0.00000000514
#> 2 ST_mel… LYPL… -0.404 1 1.60e- 9 1.68e- 8 3.36e-9 0.0000000110
#> 3 ST_mel… PCNA -0.746 1 0 0 1.79e-8 0.0000000546
#> 4 ST_mel… METT… -0.730 1 0 0 3.73e-7 0.000000912
#> 5 ST_mel… FASN -0.631 1 8.95e-10 9.69e- 9 1.90e-6 0.00000382
#> 6 ST_mel… RPS2… -0.739 1 3.80e-14 6.82e-13 5.90e-6 0.0000116
#> 7 ST_mel… EPHA2 -0.466 1 3.22e-12 4.53e-11 1.12e-5 0.0000213
#> 8 ST_mel… IVNS… -0.683 1 3.89e-14 6.86e-13 1.64e-5 0.0000305
#> 9 ST_mel… B2M -0.426 1 9.20e- 8 7.67e- 7 2.07e-5 0.0000381
#> 10 ST_mel… PRAME -0.656 1 4.55e-12 6.29e-11 2.16e-5 0.0000391
#> # ℹ 2,990 more rows
#> # ℹ 1 more variable: comments <chr>The columns p_val and adj_p_val are
respectively the nominal and adjusted p-values resulting from the
differential expression tests using the non-spatial models. The
exp_p_val and exp_adj_p_val are the p-values
resulting from the spatial models.
Comparing results from the non-spatial and spatial models
We can look at the -log10(p-values) to compare each spatial model to the non-spatial model:
# First load ggrepel to add gene names to plot
library('ggrepel')
# Calculate -log10 p-values
log_pval <- deg[['ST_mel1_rep2']] %>%
select(gene, cluster_1, mm_p_val, exp_p_val) %>%
mutate(log_p_val= -log10(mm_p_val+1e-100)) %>% # Add a constant to avoid -log10(0)
mutate(log_exp_p_val= -log10(exp_p_val+1e-100))
# Create plot
p1 <- ggplot(log_pval, aes(x=log_p_val, y=log_exp_p_val)) +
geom_abline(intercept=0, slope=1, linetype="dashed") + # Diagonal line... All p-values over this line if equal
geom_hline(yintercept=-log10(0.05), linetype="dashed", color='gray50') + # Line p-value <0.05
geom_vline(xintercept=-log10(0.05), linetype="dashed", color='gray50') + # Line p-value <0.05
geom_point(aes(color=cluster_1)) +
xlim(0, 20) + ylim(0, 20) + # Restrict axes to ease visualization (very small p-values)
geom_text_repel(aes(label=gene), size=2, show.legend=FALSE) +
theme_bw()
p1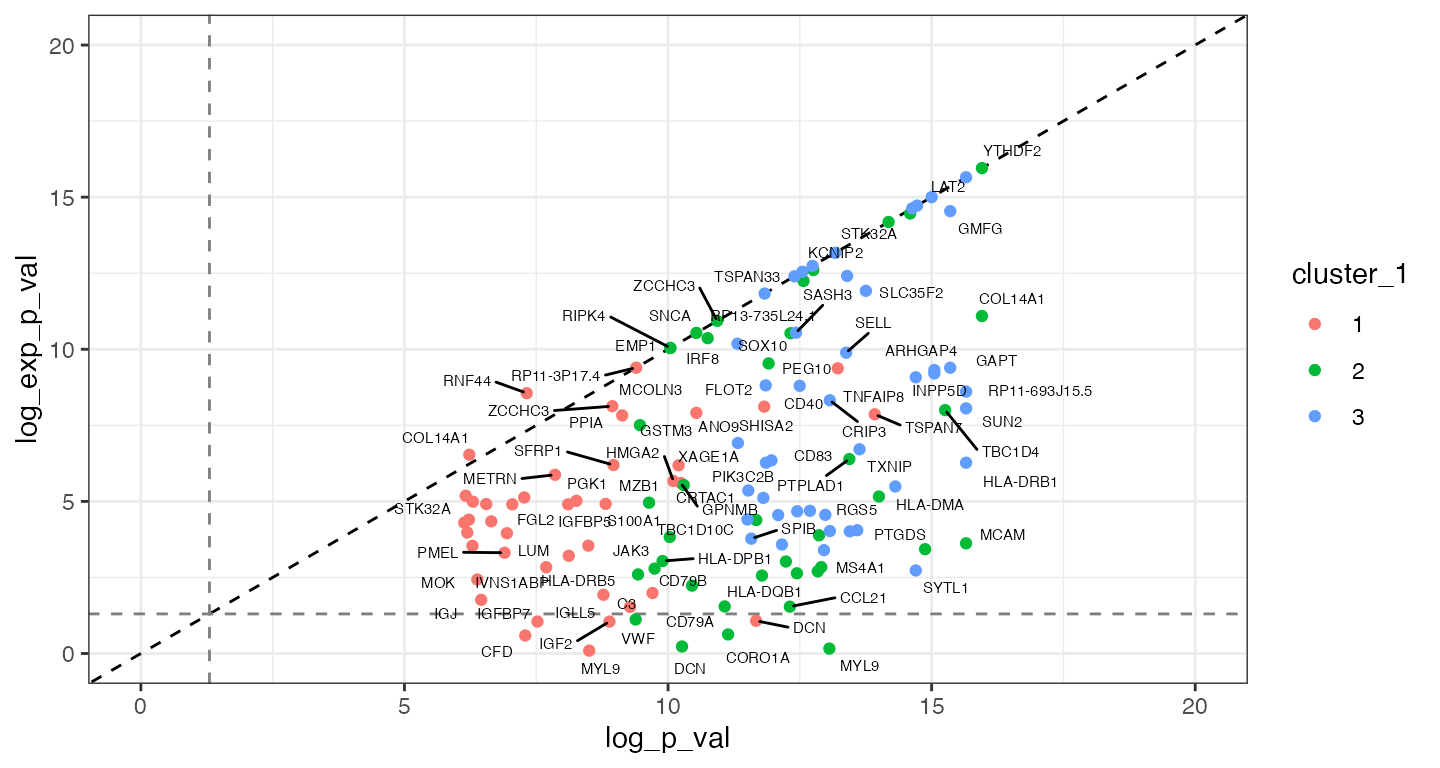
When comparing the -log10(p-values) from the non-spatial and spatial models, it is possible to see that the p-values from the non-spatial model tended to provide smaller, more significant p-values (i.e., larger -log10(p-values), below the 1:1 dotted red line). Some genes such as DCN, MYL9, IGF2 are shown as differentially expressed by the non-spatial models, however, are not differentially expressed in the spatial models (p-values under the -log10(0.05) indicated with horizontal and vertical dashed lines). This pattern is suggestive of type I error inflation.
Testing spatial models with STdiff is both
computationally- and time-intensive. The analysis in this tutorial took
approximately 18 minutes in a OS machine with a 6-cores Intel Core i7
and 16GB memory. Should the user decide to disable spatial testing, the
user only needs to set sp_topgenes=0. When running
spatial tests with more genes, and samples assayed with more
densely sampled technologies (e.g., Visium, CosMx-SMI), the user should
consider running STdiff in a High-Performance Computing
environment (HPC, or “cluster”), setting the number of cores to use via
the cores parameter.
Does spatialGE support other differential
expression test types?
It has been shown above that accounting for spatial dependence
between spots/cells is important to control inflation of type-I error
(i.e., too many differentially expressed genes). Nonetheless, the
STdiff function also implements differential expression
analysis using tests commonly used in non-spatial single cell analysis.
Specifically, the argument test_type can be set to
“wilcoxon” or “t_test” to perform either Wilcoxon’s Rank tests or
T-tests respectively:
deg_wilcox <- STdiff(melanoma,
samples='ST_mel1_rep2',
w=0.025, k='dtc', deepSplit=FALSE,
topgenes=1000,
test_type='wilcoxon', cores=2)
#> Testing STclust assignment (w=0.025, dtc deepSplit=False)...
#> Running Wilcoxon's tests...
#> Completed Wilcoxon's tests (0.46 min).
#> STdiff completed in 0.46 min.No spatial tests are executed when Wilcoxon’s Rank tests or T-tests are requested.
print(deg_wilcox[['ST_mel1_rep2']])
#> # A tibble: 3,000 × 6
#> sample gene avg_log2fc cluster_1 wilcox_p_val adj_p_val
#> <chr> <chr> <dbl> <chr> <dbl> <dbl>
#> 1 ST_mel1_rep2 IGLL5 1.03 1 1.22e-15 3.90e-14
#> 2 ST_mel1_rep2 PCNA -0.746 1 1.66e-15 4.99e-14
#> 3 ST_mel1_rep2 GNAS -0.715 1 5.08e-15 1.34e-13
#> 4 ST_mel1_rep2 COL1A1 1.06 1 5.77e-14 1.24e-12
#> 5 ST_mel1_rep2 METTL9 -0.730 1 1.51e-13 3.10e-12
#> 6 ST_mel1_rep2 CPEB2 -0.579 1 1.45e-12 2.56e-11
#> 7 ST_mel1_rep2 SSFA2 -0.612 1 2.05e-12 3.57e-11
#> 8 ST_mel1_rep2 EPHA2 -0.466 1 4.10e-12 6.94e-11
#> 9 ST_mel1_rep2 STK32A -0.451 1 9.61e-12 1.53e-10
#> 10 ST_mel1_rep2 PEG10 -0.469 1 1.62e-11 2.46e-10
#> # ℹ 2,990 more rowsVisualizng STdiff results
The spatialGE package has a built-in function
(STdiff_volcano) to generate volcano plots. The function
takes STdiff outputs and generates a volcano plot for each
sample and cluster (or cluster pair if pairwise testing was
requested).
vp <- STdiff_volcano(deg)Now, the ggpubr package can be used to arrange all
plots:
ggpubr::ggarrange(plotlist=vp, ncol=3, common.legend=TRUE)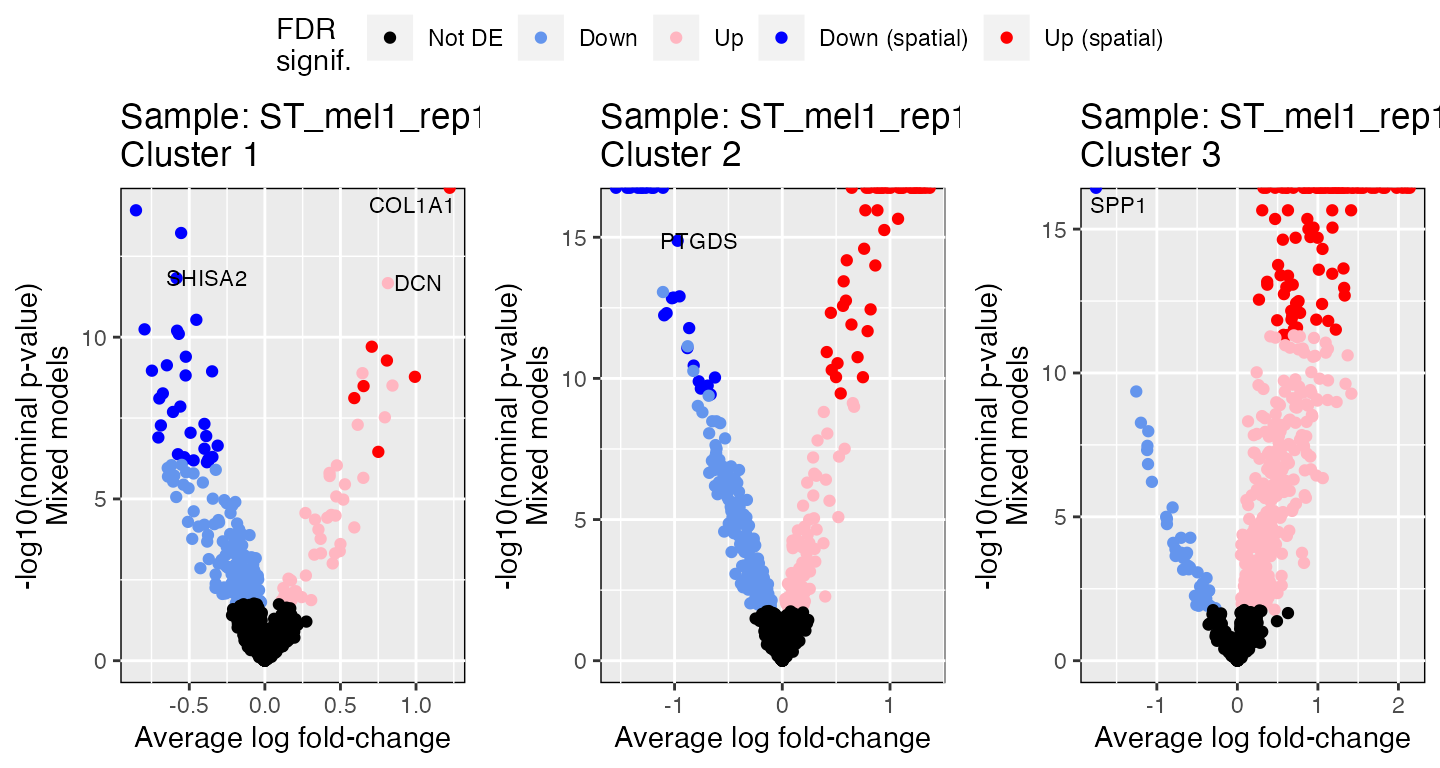
References
Session Info
sessionInfo()
#> R version 4.4.2 (2024-10-31)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS Sequoia 15.3.2
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: America/New_York
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggrepel_0.9.5 lubridate_1.9.3 forcats_1.0.0 stringr_1.5.1
#> [5] dplyr_1.1.4 purrr_1.0.2 readr_2.1.5 tidyr_1.3.1
#> [9] tibble_3.2.1 ggplot2_3.5.1 tidyverse_2.0.0 spatialGE_1.2.0
#>
#> loaded via a namespace (and not attached):
#> [1] tidyselect_1.2.1 khroma_1.13.0 farver_2.1.2
#> [4] fastmap_1.2.0 digest_0.6.37 timechange_0.3.0
#> [7] lifecycle_1.0.4 magrittr_2.0.3 compiler_4.4.2
#> [10] rlang_1.1.4 sass_0.4.9 tools_4.4.2
#> [13] utf8_1.2.4 yaml_2.3.10 ggsignif_0.6.4
#> [16] knitr_1.48 labeling_0.4.3 htmlwidgets_1.6.4
#> [19] bit_4.0.5 sp_2.1-4 RColorBrewer_1.1-3
#> [22] abind_1.4-5 registry_0.5-1 withr_3.0.1
#> [25] numDeriv_2016.8-1.1 desc_1.4.3 grid_4.4.2
#> [28] fansi_1.0.6 ggpubr_0.6.0 colorspace_2.1-1
#> [31] scales_1.3.0 MASS_7.3-61 ggpolypath_0.3.0
#> [34] cli_3.6.3 rmarkdown_2.28 crayon_1.5.3
#> [37] ragg_1.3.2 generics_0.1.3 rstudioapi_0.16.0
#> [40] tzdb_0.4.0 minqa_1.2.8 pbapply_1.7-2
#> [43] cachem_1.1.0 proxy_0.4-27 parallel_4.4.2
#> [46] vctrs_0.6.5 boot_1.3-31 Matrix_1.7-1
#> [49] carData_3.0-5 jsonlite_1.8.8 slam_0.1-52
#> [52] spaMM_4.5.0 car_3.1-2 hms_1.1.3
#> [55] rstatix_0.7.2 bit64_4.0.5 systemfonts_1.1.0
#> [58] jquerylib_0.1.4 splancs_2.01-45 glue_1.7.0
#> [61] ROI_1.0-1 pkgdown_2.1.1 nloptr_2.1.1
#> [64] cowplot_1.1.3 stringi_1.8.4 gtable_0.3.5
#> [67] munsell_0.5.1 pillar_1.9.0 htmltools_0.5.8.1
#> [70] R6_2.5.1 tcltk_4.4.2 textshaping_0.4.0
#> [73] vroom_1.6.5 evaluate_0.24.0 lattice_0.22-6
#> [76] highr_0.11 backports_1.5.0 broom_1.0.6
#> [79] bslib_0.8.0 Rcpp_1.0.13 gridExtra_2.3
#> [82] nlme_3.1-166 checkmate_2.3.2 geoR_1.9-4
#> [85] xfun_0.47 fs_1.6.4 pkgconfig_2.0.3